Cystisk fibrose
Cystisk fibrose er en alvorlig arvelig sygdom, som hovedsagelig påvirker lungerne, bugspytkirtel, men kan involvere andre organer. Symptomerne begynder normalt i den tidlige barndom og omfatter vedvarende hoste, hvæsen, gentagne lungeinfektioner, malabsorption af mad og generelt dårligt helbred. Behandlinger omfatter antibiotika, fysioterapi, slim udtynding medicin, pancreas enzym udskiftninger og andre behandlinger.
Hvad er cystisk fibrose?

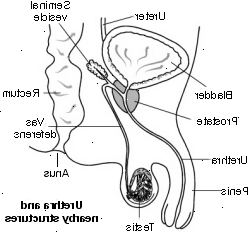
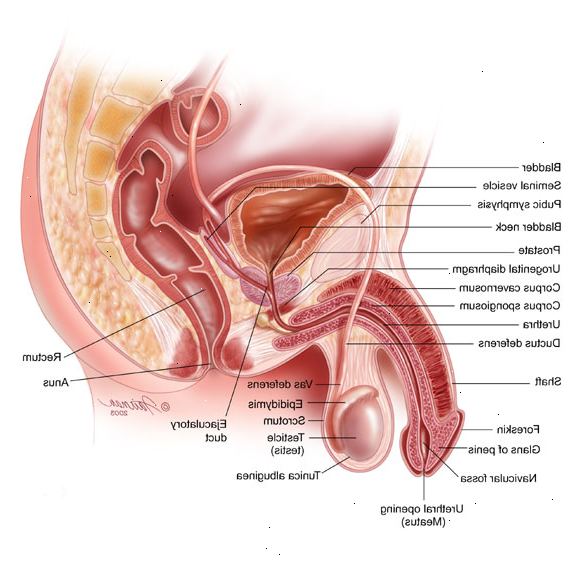
Cystisk fibrose er en tilstand, som primært påvirker lungerne, bugspytkirtel, men kan påvirke andre dele af kroppen, herunder leveren, næse og bihuler, kønsorganer, og svedkirtlerne. Normalt celler i disse dele af kroppen gør slim og andre våde safter og sekreter.
Hos mennesker med cystisk fibrose, har disse celler ikke fungere korrekt og gøre slim og sekreter, der er tykkere end normalt. Dette kan forårsage forskellige symptomer og problemer (som er beskrevet nedenfor).
Hvad er årsagen til cystisk fibrose, og hvor almindeligt er det?
Cystisk fibrose er en genetisk sygdom. Hvis du har cystisk fibrose, er en af dine gener ikke fungerer korrekt. Dette er kendt som en CFTR-genet, der er på kromosom 7.. Der er forskellige mutationer, der kan forekomme i dette gen, og dette betyder, at der er forskellige sværhedsgrader af cystisk fibrose, der opstår.
Dette gen hjælper med at styre den måde cellerne håndterer natrium-og chloridioner (salt). Mutationen i genet resulterer i cellerne ikke er i stand til at håndtere natrium og klorid ordentligt.
Som en følge heraf har celler i de berørte organer en fejl i den måde natrium og klorid rejser i og ud af cellerne. Dybest set, for meget natrium rejser i cellerne. Vand følger natrium, hvilket efterlader for lidt vand uden for cellerne. Dette bevirker, at slim eller våde sekreter uden for cellerne at være for tyk (for eksempel i luftvejene i lungerne).
Omkring 1 i 2.500 babyer i UK er født med cystisk fibrose. Over 9.000 mennesker har i øjeblikket cystisk fibrose i Storbritannien. Cystisk fibrose er en autosomal recessiv sygdom. Det betyder, at for at udvikle cystisk fibrose du nødt til at arve to cystisk fibrose gener, en fra din mor og en fra din far. Hvis du arver kun en cystisk fibrose-genet, er du kaldes et luftfartsselskab.
Omkring 1 i 25 mennesker i Storbritannien af kaukasisk afstamning (hvid European) er bærere af genet for cystisk fibrose. Det er langt mindre udbredt i afro-caribiske og asiatiske mennesker. Luftfartsselskaber ikke har sygdommen, da de har et normalt gen, som kan styre salt transport i deres celler. Men luftfartsselskaber kan passere genet for cystisk fibrose videre til deres børn.
Når to mennesker, der bærer genet for cystisk fibrose har et barn, der er en:
- 1 ud af 4 chance for, at barnet vil have cystisk fibrose (ved at arve cystisk fibrose genet fra begge forældre).
- 2 i 4 chance for, at barnet ikke vil have cystisk fibrose, men vil være en bærer (ved arve en cystisk fibrose-genet fra den ene forælder, men det normale gen formular den anden forælder).
- 1 i 4 chance for, at barnet ikke vil have cystisk fibrose, og vil ikke være en transportør (ved arve det normale gen fra begge forældre).
Hvad er symptomerne på cystisk fibrose?
Cystisk fibrose symptomer normalt først udvikle sig inden for det første år af livet, men kan ikke vises, før senere barndom. Sværhedsgraden af symptomerne kan variere.
Lungesymptomer

De celler, der linje luftvejene i lungerne gør sputum (slim), der er tykkere end normalt, og som ikke er frigivet fra lungerne nemt. Dette kan fælde bakterier i de små luftveje og føre til infektion og inflammation. Så symptomer, som typisk udvikler omfatter:
- Vedvarende hoste der typisk producerer en masse spyt.
- Hvæsende vejrtrækning.
- Åndenød og åndedrætsbesvær.
- Tilbagevendende lungeinfektioner. Disse kan være alvorlige, såsom lungebetændelse. Gentagne infektioner og betændelse kan beskadige lungerne og føre til dårlig lungefunktion.
Gut symptomer
Bugspytkirtlen normalt gør mavesyren, der indeholder kemikalier (enzymer). Mavesyren normalt strømme ud fra pancreas kanalen ind i duodenum og fordøje maden.
Hos mennesker med cystisk fibrose, blokere fortykket sekret den normale strøm af mavesyren fra bugspytkirtlen. Dette kan resultere i fødevarer ikke bliver fordøjet eller absorberet ordentligt - især fed mad og fedtopløselige vitaminer (vitamin A, D, E og K).
Dette kan forårsage:
- Fejlernæring fører til dårlig vækst og dårlig vægtøgning (selvom du har en god appetit og spiser en masse, da problemet er med at fordøje og absorbere fødevarer).
- Store, ildelugtende, fedtet, fede fæces (afføring eller bevægelser), forekommer i omkring en tredjedel af tilfældene.
- Oppustet mave.
- Forstoppelse.
I omkring 3 ud af 10 tilfælde bugspytkirtel fungerer godt, og der er ingen eller minimal gut symptomer, og først og fremmest bare lungesymptomer.
Symptomer undertiden forekomme ved fødslen

Omkring 1 ud af 10 børn med cystisk fibrose er diagnosticeret kort efter fødslen. Dette skyldes en tilstand kaldet mekonium ileus hvor der i nogle tilfælde tarmen bliver blokeret med mekonium - en tyk, mørk, klæbrig substans som er lavet af barnets tarmen før de fødes. Presserende kirurgi kan være nødvendig for at lindre blokering.
Andre symptomer og komplikationer
Andre organer kan blive påvirket som kan forårsage forskellige andre problemer i nogle tilfælde. Desuden kan bugspytkirtlen og luftveje bliver hårdt ramt. Derfor, andre problemer, der kan også forekomme i nogle tilfælde omfatter:
- Gentagen sinus infektioner.
- Polypper dannes i næsen.
- Ufrugtbarhed (især hos hanner, kan de rør, der fører sæden bliver blokeret).
- Skade på leveren, hvilket kan føre til skrumpelever, forekommer i omkring 1 i 12 tilfælde (hvis de små kanaler i leveren bliver blokeret eller beskadiget).
- Diabetes. (Særlige celler i bugspytkirtlen at gøre insulin. Hvis bugspytkirtlen bliver alvorligt beskadiget over tid derefter insulin niveauer gå ned og diabetes kan udvikle sig.) Dette er sjælden hos børn, men er mere almindelig hos voksne, der har haft cystisk fibrose.
- Pancreatitis (betændelse i bugspytkirtlen).
- Rektal prolaps.
- Osteoporose (udtynding af knoglerne) kan udvikles på grund af dårlig absorption af visse fødevarer, og i særdeleshed, som D-vitamin er nødvendig for at opretholde sunde knogler.
- Den sved smager meget salt.
Milde tilfælde
Nogle tilfælde af cystisk fibrose er diagnosticeret hos voksne, der har relativt milde symptomer. Dette kan være på grund af nogle mutationer i genet for cystisk fibrose ikke er så defekt som andre. Håndteringen af natrium og klorid kan kun svagt påvirket i disse tilfælde.
Hvordan cystisk fibrose diagnosticeres?
Sweat test
En læge kan arrangere en sved test, hvis han eller hun mistænker cystisk fibrose fra symptomerne. Denne test måler mængden af salt (natrium og chlorid) i huden sved. Mennesker med cystisk fibrose har et unormalt højt saltindhold i sved.
Genetisk test
En genetisk test kan bekræfte diagnosen. Nogle celler er enten skrabet fra indersiden af kinden eller taget fra en blodprøve. Disse kan afprøves for at detektere genet for cystisk fibrose.
Screening test

Alle nyfødte i Storbritannien er nu screenet for cystisk fibrose. En lille hæl pik blodprøve er taget ved den sjette dag efter fødslen. Dette kan registrere et kemisk stof kaldet immunoreaktiv trypsinogen, som er høj i babyer med cystisk fibrose. Hvis det er højt, en svedtesten og genetisk test kan gøres for at bekræfte diagnosen. Screening anses for vigtigt, da den tidligere diagnosen stilles, jo før kan behandlingen begynde som forbedrer udsigterne (prognose).
Cystisk fibrose behandling
Der er mange aspekter til behandling af mennesker med cystisk fibrose. Behandlingen involverer input, rådgivning og ekspertise fra forskellige fagfolk, såsom børns sundhed læger, specialsygeplejersker, fysioterapeuter, diætister, rådgivere og psykologer samt din primære sundhedspleje team.
Det er normalt at have regelmæssige kontroller og prøver at overvåge tilstanden og for at holde styr på børns vækst, udvikling og trivsel.
Følgende liste er en kort oversigt over de almindeligt anvendte behandlinger, men er ikke en fuld eller udtømmende redegørelse for alle anvendte behandlinger. En individuel behandlingsplan er nødvendig for hver enkelt sag at tage hensyn til individuelle omstændigheder.
Behandlinger for lungeproblemer
Fysioterapi og motion
Regelmæssig lungefysioterapi er meget vigtigt. Dette hjælper med at rydde luftvejene af den tykke slim. En fysioterapeut viser normalt forældrene, hvordan at gøre dette til deres børn. Det indebærer en særlig måde at fast klappe brystet, mens barnet ligger med hovedet nedad for at fremme slim og spyt skal hostede ud. To gange dagligt lungefysioterapi er almindelig praksis. Dette kan være nødvendigt at øge i tider med lungeinfektioner.
Der er en bred vifte af luftvejs clearance teknikker til rådighed, og din fysioterapeut vil være i stand til at rådgive dig om den mest egnede teknik eller teknikker for dig. Disse vil sandsynligvis ændre sig selv, da du bliver ældre, eller som din sygdom ændrer sig med tiden.
Det er også vigtigt at opfordre børn til at udøve og til at være så aktiv og passer som muligt. Så sport og spil såsom løb, svømning, fodbold og tennis fremmes.
Antibiotika og antisvampemidler
Kurser af antibiotika er en grundpille i behandling. Mange børn med cystisk fibrose tager regelmæssig langsigtet antibiotika. Dosis øges og / eller andre typer af antibiotika er givet, når en kiste infektion udvikler sig. Forskellige bakterier kan forårsage infektioner og de valgte antibiotika afhænger af, hvilke bakterier er fundet i prøver af spyt. Antibiotika gives intravenøst (i en vene) er ofte påkrævet for alvorlige infektioner, der ikke kan kontrolleres med antibiotika tabletter. De kan også gives af en forstøver.
En bakterie kaldet Pseudomonas aeruginosa almindeligvis fortsætter i tykke slim i luftvejene. For at holde denne blusser op i gentagne infektioner, et antibiotikum givet af nebulisator (inhaleret antibiotika), eller inhalator er en fælles behandling.
Undertiden lungerne bliver inficeret med en svamp og svampedræbende medicin er nødvendig.
Inhalatorer
Inhalatorer at åbne luftvejene så meget som muligt, kan anvendes - for eksempel salbutamol. Dette svarer til den behandling, der anvendes til astma.
Dornase alfa
Dette er et lægemiddel afgivet nebulisator i nogle tilfælde. Det hjælper til at nedbryde og til at fortynde tykke slim, hvilket gør det lettere at hoste op og rydde slim fra luftvejene. Det kan reducere antallet af lungeinfektioner og bidrage til at forbedre lungefunktionen.
Oxygen
Mennesker med fremskreden lungesygdom kan drage fordel af ilt, især natten over.
Anden medicin til at forbedre lungefunktionen - for eksempel, og ibuprofen azithromycin - kan også anbefales i nogle tilfælde.
BEHANDLINGER for pancreas PROBLEMER
Ernæring
De enzymer, der er nødvendige for at fordøje maden er stærkt reduceret i de fleste mennesker med cystisk fibrose. Derfor børn med cystisk fibrose brug for en høj fedt og kulhydrat kost. En diætist vil normalt give detaljerede råd. Høj-energi drikke kosttilskud kan også være nødvendig. Desuden er vitamintilskud nødvendig, da mange vitaminer i fødevarer ikke absorberes meget godt. Bliver velnærede vil også hjælpe dig til at bekæmpe enhver lungeinfektioner.
Enzym kosttilskud
I de fleste tilfælde er enzymtilskud nødvendig for at hjælpe til at fordøje maden. (Disse erstatter de enzymer, der normalt kommer fra bugspytkirtlen.) Du er nødt til at tage disse kosttilskud, hver gang du spiser mad. Dette kan betyde, at mange doser hver dag.
Andre behandlinger
En række andre problemer, der er relateret til cystisk fibrose, kan udvikle sig i nogle tilfælde og kræver behandling. For eksempel:
- Saltmangel kan forekomme i varmt vejr og kan kræve salt kosttilskud.
- Leverproblemer udvikle sig i nogle tilfælde og kan kræve specialiserede liver behandlinger.
- Hvis diabetes udvikler sig, det normalt kræver insulin eller tabletbehandling.
- Nasale polypper sommetider udvikler og kan behandles med steroid næsedråber og sprays.
- Acid reflux fra maven i spiserøret (spiserøret) er fælles og kan behandles med medicin, der nedsætter syreindholdet i maven saft.
- Forstoppelse er ret almindelige og kan kræve regelmæssige afføringsmidler.
- Alle mennesker med cystisk fibrose bør være up to date med rutinemæssige vaccinationer, og har også en årlig influenza jab til at forebygge influenza og et pneumokokvaccine til at forhindre lungebetændelse forårsaget af denne bakterie.
- Nogle børn bliver testet for deres immunitet mod skoldkopper (skoldkopper). Hvis du ikke har immunitet over for det, så du kan blive tilbudt vaccinen mod skoldkopper.
- Lunge eller hjerte / lunge transplantation kan tilbydes i nogle tilfælde som lungesygdom bliver mere alvorlige.
Nyere behandlinger bliver forsket og udviklet, og hvis fundet en succes, kan blive mere udbredt i fremtiden. For eksempel:
- Genterapi. Dette indebærer anvendelse af en inhaleret spray til at levere normale kopier af genet for cystisk fibrose til lungerne.
- Lægemidler testes som kan korrigere den abnorme salt og vand regulering af celler, der fører til fortykket slim og sekreter, der gøres i lungerne og andre organer.
- Nye metoder til at forbedre virkningen af de nuværende behandlinger er under udvikling.
Hvad er udsigterne (prognose)?
Cystisk fibrose er en livslang tilstand. Med forbedret behandling har der været en dramatisk stigning i overlevelse af mennesker med cystisk fibrose over en periode på 20 år eller deromkring.
I 1960'erne og før, de fleste babyer født med cystisk fibrose kun overlevede i et par måneder eller år. I dag er mange mennesker med cystisk fibrose lever i deres sene 30'erne og videre. Med optimal pleje og behandling, skønnes det, at omkring 8 ud af 10 af nutidens børn med cystisk fibrose skal leve i deres midten af 40'erne eller 50'erne. Med behandling, kan de fleste mennesker med cystisk fibrose lever rimeligt normalt og produktivt liv.
Dog vil der være tidspunkter, hvor symptomerne er mere alvorlige - hovedsageligt når en kiste infektion udvikler sig. Selv med behandling, er den største risiko tilbagevendende lungeinfektioner og lungebetændelse. Dette kan have en gentagen skadelig virkning på lungefunktionen, som kan blive værre med tiden. Døden i barndommen eller den tidlige voksenalder er stadig ikke ualmindeligt. De fleste mennesker med cystisk fibrose dør af lunge komplikationer, primært respiratoriske, og hjertesvigt.
Genetisk rådgivning
Mennesker med en familie historie af cystisk fibrose kan ønske at have genetisk rådgivning og testning for at finde ud af deres risiko for at overføre betingelsen videre til deres børn. En simpel test kan gøres for at se på de gener fra celler fra indersiden af kinden eller fra blod. Testen kan detektere genet for cystisk fibrose, som kan vise, om man er bærer af den abnorme gen.
Yderligere hjælp og information
Cystisk fibrose tillid
11 London Road, Bromley, Kent, BR1 1BY
Tlf (helpline): 0845 859 1000 Web: www.cftrust.org.uk
Giver information og støtte til mennesker med cystisk fibrose og deres plejere.
Cystisk fibrose screeningsprogram
Web: www.screening.nhs.uk / cysticfibrosis-nyfødte
Oplysninger om det nationale screeningsprogram for cystisk fibrose hos den nyfødte.